Browse through our Journals...
DEVELOPMENT, EVALUATION AND METHOD SELECTION FOR THE PREPARATION OF LAMIVUDINE MICROSPHERES
AMITAVA GHOSH*, UDAYA KUMAR NAYAK AND PARTHA ROY
Department of Pharmaceutics
Himalayan Pharmacy Institute, Majhitar, East Sikkim, India -737136
ABSTRACT
The present study concerns the preparation, evaluation and selection of a suitable for the preparation of lamivudine incorporated microspheres composed of ethyl cellulose as release controlling polymeric material. Microspheres were prepared from various methods, namely, modified w/o/o emulsion solvent evaporation method (F1), o/w/o type emulsion solvent evaporation method (F2), thermal change technique (F3), Nobel Quassi emulsion solvent diffusion method (F4), meltable dispersion method (F5) , phase separation coacervation method (non-solvent addition technique) (F6). The prepared microspheres were evaluated for parameters such as, percentage yield, drug entrapment efficiency, particle size determination, drug polymer interaction, stability studies, and invitro drug release kinetic study. The drug / polymer ratio (1:2 w/w) and drug load (100 mg), was kept constant throughout the current investigation. The microspheres produced in each batch were smooth and small in size with an average diameter about 27.89 - 41.28 µm. Microspheres exhibited high drug entrapment efficiency with high yield value. FTIR analysis confirmed the absence of drug polymer interactions in all the formulations. The accelerated stability studies were performed following ICH guidelines for 14 Weeks and the results indicated stability of the formulations in varying temperature. Drug release profile of microspheres from F1, F2 and F3 followed zero order kinetics while those from F4, F5 and F6 formulation fit to Higuchi square root model. Among the methods adopted in this study, thermal change method (F3) was most successful in sustaining the release of lamivudine from ethyl cellulose microspheres, a novel trend for effective management of AIDS.
KEYWORDS: Lamivudine, Ethylcellulose, Microspheres, Thermal Change Method.
INTRODUCTION
New drug delivery technologies are revolutionizing the drug discovery, development and creating R&D focused pharmaceutical industries to increase the momentum of global advancements. In this regard novel drug delivery systems (NDDS) have many benefits, which includes improved therapy by increasing the efficacy and duration of drug activity, increased patient compliance through decreased dosing frequency and convenient routes of adminstration and improved site specific delivery to reduce unwanted adverse effects (Amrita Bajaj et al, 2006). Lamivudine is an active antiretroviral drug belonging to non-nucleosides reverse transcriptase inhibitor. Lamivudine treatment has gained immense popularity in the AIDS treatment in the present era. Dosage and duration of lamivudine therapy should be individualized according to requirement and response of the patient. The daily recommended dose is 150 mg b.i.d (J.H. Kao et al, 2000) (Indian Pharmacoepia, 1996). The oral administration of lamivudine exhibits side effects in GIT as well as in CNS. Thrombocytopenia, parasthesias, anorexia, nausea, abdominal cramps, depressive disorders, cough and skin rashes etc have also been reported as possible adverse reactions (Caroline M. Perry et al, 1997). Controlled release (CR) preparations helps to achieve maximum therapeutic effect with simultaneous minimization of adverse effects. Microparticulate drug delivery posses many advantages such as high bioavailability, rapid kinetic of absorption as well as avoidance of hepatic first pass effect and improvement of patient compliance (Y.W. Chien, 1992). Absence of sufficient work in the direction of programmed delivery of lamivudine as indicated by literature survey ignited the urge of this research venture, which utilizes six different formulation methods for preparation of lamivudine microspheres and ultimately ascertain the most preferable method for industrial scale-up on the basis of physical characterization and in vitro drug release profile.
METHODS AND AIMS
Materials
Lamivudine was received as a gift sample from GlaxoSmithKline Ltd. Mumbai. Ethyl cellulose was obtained from LOBA chemicals, Kolkata, India. All other chemicals and solvents used were of analytical grade and procured from an authorized dealer, USP XXI paddle type dissolution apparatus, FT-IR (Shimadzu IR spectrophotometer, Model 840, Japan) and UV-Visible spectrophotometer (UV-1700, Shimadzu, Japan) were the instruments employed in the current study.
Preparation of microspheres
The Lamivudine loaded Ethyl cellulose microsphere were prepared by modified w/o/o emulsion solvent evaporation method (F1) (Y. Li et al , 2005), o/w/o type emulsion solvent evaporation method (F2) (D. H. Kenneth et al, 2005), thermal change technique (F3) (C. Palanichamy et al, 2006), Nobel Quasi emulsion solvent diffusion method (F4) (Y. Kawashima et al, 1989), meltable dispersion method (F5) (J.B. Schwartz et al 1968), phase separation coacervation method (non-solvent Addition Technique) (F6) (K.N.Shovarani et al, 1994), as described in referred literatures. The ratio of drug and polymer concentration (1:2 w/w) along with drug load (100 mg) was kept constant throughout the study period.
Drug entrapment efficiency (DEE) (M.C. Gohel et al, 2005)
The microspheres were evaluated for percentage yield and percent drug entrapment. The yield was calculated
Percentage yield = weight of microsphere recovered X 100
Weight (drug + polymer)
…………. (1)
Drug loaded microspheres (100 mg) were powdered and suspended in 100 ml methanol: water (1:99 v/v) solvent system. The resultant dispersion was kept for 20 min for complete mixing with continuous agitation and filtered through a 0.45 µm membrane filter. The drug content was determined spectrophotometrically (UV-Visible-1700, Shimadzu, Japan spectrophotometer) at 270 nm (3) using a regression equation derived from the standard graph (r2 = 0.9978). The drug entrapment efficiency (DEE) was calculated by the equation
DEE = (Pc / Tc) X 100
…………….. (2)
Pc is practical content, Tc is the theoretical content. All the formulations were analyzed in triplicate (n=3).
Particle size measurement (M.C. Gohel et al, 2005)
The size of the prepared microspheres was measured by the optical microscopy method using a calibrated stage micrometer. Particle size was calculated by using equation
Xg = 10 x [(ni x log Xi) / N]
......……………….. (3)
Xg is geometric mean diameter, ni is number of particle in range, xi is the mid point of range and N is the total number of particles. All the experimental units were analyzed in triplicate (n=3).
Accelerated stability studies (G.T. Kulkarni et al, 2004) (L. Lachman, et al, 3rd ed., 1991)
Stability studies were performed according to ICH guidelines. The formulations were stored in room temperature at 25 ± 1°, in hot air oven at 37 ± 1°, and at 60± 1° for a period of 14 weeks. The samples were analyzed for drug content every two weeks by spectrophotometer at 270 nm and compatibility of drug with excipients was determined by infrared spectroscopy using a Shimadzu FTIR-840 model IR spectrophotometer.
Fourier Transforms Infrared Radiation measurement (FT-IR) (D.R.Bhumkar et al, 2003)
The FT-IR spectra acquired were taken from dried samples. A FT-IR (Shimadzu IR spectrophotometer, model 840, Japan) was used for the analysis in the frequency range between 4000 and 600 cm-1, an 8 cm-1 resolution and a 0.2 cm-1 rate. The results were the means of 16 determinations. A quantity equivalent to 2 mg of pure drug, empty microspheres of ethyl cellulose and drug loaded microspheres were selected separately.
In-vitro drug release
Invitro drug release study was carried out in USP XXI paddle type dissolution test apparatus using 0.01 M HCl as dissolution medium. Volume of dissolution medium was 900 ml and bath temperature was maintained at 37±1° throughout the study. Paddle speed was adjusted to 50 rpm. At an interval of 1 hr, 5 ml of sample was withdrawn with replacement of 5 ml fresh medium and analyzed for lamivudine content by UV-Visible spectrophotometer at 270 nm (3). All the experimental units were analyzed in triplicate (n=3).
In vitro drug release kinetics
In order to study the exact mechanism of drug release from the microsphere, drug release data was analyzed according to Zero order (G.M. Khan, 2001), First order (D.M. Morkhade et al, 2006), Higuchi square root (T. Higuchi, 1963), Hixon-Crowell equation (J. Wang et al, 1999) .The criteria for selecting the most appropriate model was chosen on the basis of goodness of fit test.
Statistical Analysis
Statistical data analyses were performed using the ANOVA one way at 5 % level of significance p < 0.05 (Bolton S, 1997) and standard error mean.
RESULTS
The Lamivudine loaded ethyl cellulose microspheres were prepared by various methods as mentioned earlier. The microspheres obtained under these conditions were found to be spherical and without aggregation and mean particle size was found in a range of 27.89 to 41.28 µm (Table 1). The particle size distribution of all the formulations is displayed in Figure-2. The percentage yield of all the formulations was found to be satisfactory and each formulation exhibited high drug entrapment efficiency (DEE), as summarized in Table 1. The method F3 showed higher DEE among all the formulations.
The interaction study between the drug (lamivudine) and polymer (ethyl cellulose) in different formulations was evaluated using FTIR spectrophotometer. Four bands present in Lamivudine spectrum at 3445.91, 2930.92, 1736.51 and 1637.7 cm-1, due to the formation of N-H, O-H, C=O, C=N linkage respectively, was also detected and identified in the spectrum of the formulations, confirming no drug-polymer interaction as represented in Figure-3, 4 and 5. The accelerated stability studies were performed according to ICH guidelines for 14 Weeks and the results were found to be stable in varying temperature as shown in Table-2. The results were further verified with one way ANOVA method, the accelerated stability test datas were found significant for F (3.395) at 5 % level of significance (p< 0.05).
The in vitro drug release profiles for all the batches were tabulated in Table-3 and graphically represented in Figure-1. All the formulations showed constant release profile. To identify the kinetics of drug release from microspheres, release data was analyzed according to different kinetic models. Table-4 indicates that drug release from F1; F2 and F3 formulations obey Zero order kinetics, while the release data of F4, F5 and F6 seems to fit best in Higuchi square root model. The release mechanism was not significantly influenced by formulation variables and was predominately diffusion controlled. Statistical verification with one way ANOVA method attested the fact that the drug release data were found significant for F (27.3731) at 5 % level of significance (p< 0.05).
DISCUSSION
Lamivudine loaded Ethyl cellulose microsphere were prepared and evaluated with different reported methods. No significant differences in particle size were found for the microspheres prepared by all the methods. Small variations may be attributed to different conditions (M.C. Gohel et al, 2005), like stirring speed and stirring time or fluctuations in temperatures. The smaller particle size can be due to application of wide range (5-800) of temperature change with constant stirring at 800 rpm. The reduced particle size helps in achieving our goal by enhancing the controlled delivery of Lamivudine. The method F 3 (C. Palanichamy et al, 2006) showed higher DEE among all the formulations. This can be justified on the basis of minimum process parameters, minimum drug solubility in the external phase and smaller particle size of the thermal change method in comparison with the other methods, leading to minimum drug loss. The minimal use of process parameters during the formulation of Lamivudine microsphere confirms the high yield of F 3.
The FTIR study attests the safety profile of the microspheres due to avoidance of drug polymer interaction. The accelerated stability study indicate the broader horizon of storage conditions complying with the ICH guidelines. The efficiency of release of Lamivudine from ethyl cellulose matrix of prepared microspheres is the key factor in the successful optimization of a method. The present study demonstrated that F3, among all other formulations, have a significantly slower release pattern in terms of their total drug load. The drug release rate was following zero order kinetic which complies with the controlled delivery (G.M. Khan, 2001) of Lamivudine over 10 hours. However the other formulations F1, F2, F4, F5 and F6, had shown insignificant difference in drug entrapment efficiency but found to be significantly (S.E.M < 0.010) distinguished in particle size and percentage of yield. Except F1, F2, F3, the other formulations followed Higuchi square root kinetic model indicating the diffusion controlled drug release, which creates a restriction in optimizing the methods, as zero order model is the most desirable. F1, F2 was rejected on the basis of particle size, yield factor, entrapment efficacy and prolongation of drug release in comparison with F3. The results of the present study suggest that method F3 is the most suited one to develop lamivudine microspheres, keeping in consideration, the zero order release profile, high DEE (99.66 %), small particle size (27.89 µm) and high yield of the microspheres of this method.
In context to the intense world wide research to combat AIDS, it can be envisaged that future workers would indulge in optimization of the various process parameters of the selected method (F3), to promote its commercial scale up, leading to Lamivudine (Caroline M. Perry et al, 1997) loaded ethyl cellulose microspheres for effective management of AIDS.
ACKNOWLEDGMENTS
The authors thank GlaxoSmithKline Pvt. Ltd. Mumbai, India for the gift sample of lamivudine pure drug and Central Drug Research Institute Lucknow, India for providing library facility during the entire work.
Table-1:
Evaluation parameters of various formulations
Formulation code |
Yield (%) |
Particle size (µm) (X ± S.D) |
Drug Entrapment efficiency ( X± S.D) |
F1
F2
F3
F4
F5
F6 |
93.00 ± 0.017
96.00 ± 0.014
99.10 ± 0.019
94.56 ± 0.027
97.55 ±0.026
98.80 ±0.023 |
36.22 ± 0.015
29.55 ± 0.021
27.89 ± 0.026
28.69 ± 0.019
38.88 ± 0.028
41.28± 0.026 |
97.01 ± 0.12
95.36 ± 0.11
99.66 ± 0.22
99.01 ± 0.19
98.45 ± 0.17
96.21 ± 0.23 |
All the results are mean ± standard deviation (n=3)
(Standard Error Mean (S.E.M) < 0.01)
F1=W/O/O Emulsion Solvent Evaporation F2= O/W/O Emulsion Solvent
Evaporation
F3= Thermal Change Method F4= Quassi emulsion solvent diffusion
Method
F5= meltable dispersion Method F6 = phase separation coacervation method
(Non-solvent addition method)
Table-2: Stability profile of various formulations in different temperature.
|
Formulation Code |
|||||||
Week |
Temp. (° C) |
F1
|
F2
|
F3
|
F4
|
F5 |
F6
|
|
Percentage of Potency |
||||||||
Initial
2
4
6
8
10
12
14 |
RoomTemperature (RT) RT 37 ± 1 60± 1
RT 37 ± 1 60± 1
RT 37 ± 1 60± 1
RT 37 ± 1 60± 1
RT 37 ± 1 60± 1
RT 37 ± 1 60± 1
RT 37 ± 1 60± 1 |
99.24
99.20 98.53 98.23
99.21 98.65 98.22
98.92 98.56 98.12
98.97 98.62 98.09
98.89 98.58 97.98
98.84 98.52 97.88
98.71 98.48 97.69 |
99.42
99.01 98.89 98.88
99.0 98.87 98.56
98.97 98.79 98.50
98.83 98.69 98.32
98.45 98.57 98.29
98.39 98.48 98.19
98.31 98.40 98.08 |
99.56
99.66 99.05 99.00
99.68 98.97 98.91
98.99 98.87 98.82
98.91 98.78 98.45
98.85 98.65 98.39
98.74 98.60 98.31
98.69 98.51 98.29 |
99.88
99.78 98.55 96.36
99.68 98.58 97.01
98.90 98.53 97.21
98.80 98.41 97.01
98.78 98.35 97.01
98.68 98.31 96.98
98.57 98.29 96.92 |
101.25
99.44 98.66 99.02
99.12 98.25 98.77
98.88 98.10 98.62
98.68 97.92 98.36
98.59 97.88 98.26
98.47 97.79 98.18
98.41 97.70 98.10 |
99.23
99.84 98.39 98.81
99.35 98.01 97.55
98.95 98.05 97.49
98.21 97.98 97.56
98.19 97.69 98.18
98.05 97.58 98.12
98.00 97.48 97.95 |
|
Verifying with one way ANOVA significant at 5 % level of significance (F= 3.395).
Table 3:
In vitro drug release profile of formulations
|
Formulation Code |
|||||
Time(hr) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
1
2
3
4
5
6
7
8
9 |
19.36
15.73
20.03
17.94
19.38
24.02
22.60
24.71
17.38 |
60.22
70.47
74.91
72.09
74.80
72.09
76.29
72.93
71.16 |
25.35
21.30
19.57
21.27
21.28
18.41
19.31
21.06
21.41 |
23.47
22.63
15.59
21.31
10.37
10.89
15.01
18.83
23.12 |
27.62
30.91
21.77
19.66
18.78
17.87
28.61
28.33
29.22 |
24.69
23.34
19.79
22.05
20.83
23.99
17.76
28.47
32.69 |
F1=W/O/O Emulsion Solvent Evaporation F2= O/W/O Emulsion Solvent
Evaporation
F3= Thermal Change Method F4= Quassi emulsion solvent diffusion
Method
F5= meltable dispersion Method F6 = phase separation coacervation method
(Non-solvent addition method)
Table-4:
Correlation coefficients according to different kinetic equations
Kinetic Models |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
( r) |
||||||
Zero order
First order
Hixon-crowell model
Higuchi square root
|
0.9992
0.7397
0.8927
0.9564
|
0.9952
0.8282
0.9443
0.9931 |
0.9891
0.8021
0.9803
0.9864 |
0.9841
0.8724
0.9727
0.9932 |
0.9968
0.8641
0.9849
0.9985 |
0.9871
0.8559
0.9853
0.9877 |
Table values represents correlation coefficient (r) for linearity according to different kinetic equations used for describing the drug release from various formulations
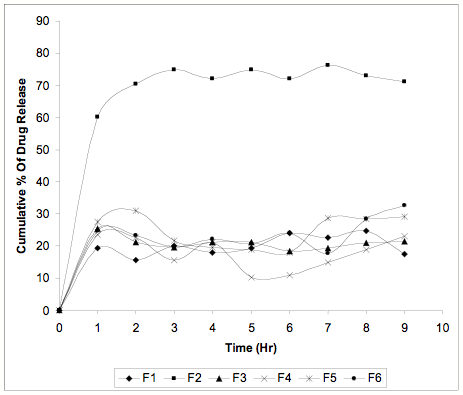
Figure-1: In vitro drug release profile of microspheres
F1=W/O/O Emulsion Solvent Evaporation F2= O/W/O Emulsion Solvent
Evaporation
F3= Thermal Change Method F4= Quassi emulsion solvent diffusion
Method
F5= meltable dispersion Method F6 = phase separation coacervation method
(Non-solvent addition method)
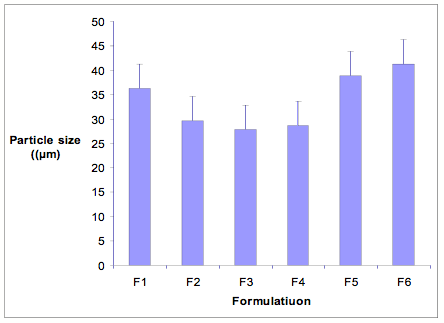
Figure-2: Particle size distribution of various formulations
F1=W/O/O Emulsion Solvent Evaporation F2= O/W/O Emulsion Solvent
Evaporation
F3= Thermal Change Method F4= Quasi emulsion solvent diffusion
Method
F5= meltable dispersion Method F6 = phase separation coacervation method
(Non-solvent addition method)
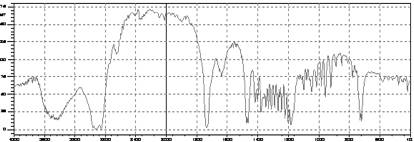
Figure-3: FTIR bands of Lamivudine pure drug
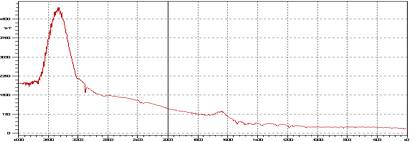
Figure-4: FTIR bands of empty microspheres of ethyl cellulose.
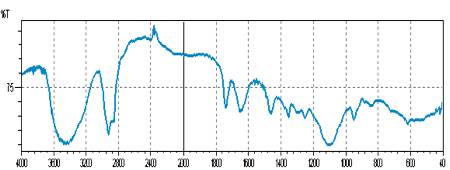
Figure-5: FTIR bands of Lamivudine loaded ethyl cellulose microsphere
REFERENCES
- Amrita Bajaj and Mansi Desai. Challenges and strategies in novel drug delivery technologies. Pharma Times, 38:12-16, 2006.
- J.H. Kao, N.H. Wu, P.J.Chen, M.Y. Lai and D.S. Chen. Hepatitis B genotype and the response to interferon therapy. J Hepatol, 33: 998-1002, 2000.
- Government of India, Ministry of Health and Family Welfare, Indian Pharmacopoeia 1996, Addendum 2002, Controller of publications, New Delhi, pp 913-914.
- Caroline M. Perry and Diana Faulds. Lamivudine- A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in the management of HIV infection. Drugs, 53: 7-680, 1997.
- Y.W.Chien, Concept and system design for rate controlled drug delivery, Novel drug delivery systems. Drug and pharmaceutical sciences, Marcel Dekker, NY, 1992.
- Y. Li, H.L Jiang, K.J. Zhu, J.H. Liu and Y.L. Hao. Preparation, Characterization and nasal delivery of α-cobrotoxin-loaded poly (lactide-co-glycolide) / polyanhydride Microspheres. J Control Release, 108: 10-20, 2005.
- D. H. Kenneth, M.C. Kathleen, M.H. Kathleen, H.L. Danny, A. P. Claude and T Schmidta. PEGylated insulin in PLGA microparticles.In vivo and in vitro analysis. J Control Release, 104: 447–460, 2005.
- C. Palanichamy, R. Priyadarsini., R. Kowsalya and S.S. Kumar. Evaluation of salbutamol sulphate microcapsules prepared by thermal change technique. Indian Drugs, 43: 96-98, 2006.
- Y. Kawashima, T. Niwa, T. Handa, H.Takeuchi, T. Iwamoto and K. Itoh. Preparation of controlled-release microspheres of ibuprofen with acrylic polymers by a novel quasi -emulsion solvent diffusion method. J Pharm Sci, 78: 68-72, 1989.
[10] J.B. Schwartz, A.P. Simonelli, and W.I. Higuchi. Drug release from wax matrices: analysis of data with first order kinetics and with diffusion controlled model. J Pharm Sci, 57:274-277, 1968.
[11] K.N.Shovarani and A.G. Goundalkar. Preparation and evaluation of microsphere of diclofenac sodium. Indian J Pharm Sciences, 56: 45-50, 1994.
[12] M.C. Gohel., R.K. Parik, A.F. Amin and A.K. Surati. Preparation and formulation optimization of sugar crosslinking gelatin microspheres of diclofenac sodium. Indian J Pharm Sci, 67: 575-581, 2005.
[13] G.T. Kulkarni, K. Gowthamarajan, and B. Suresh. Stability testing of pharmaceutical products: an over view. Indian J Pharm Educ Res, 38: 194-202, 2004.
[14] L. Lachman., P. Deluca, M.J.Akers. Kinetic principle and stability testing, in Lachman L: Lieberman H A: Kanig J L (eds), The Theory and Practice of Industrial Pharmacy. 3rd ed., Verghese Publishing House, Mumbai, 760-804, 1991.
[15] D.R. Bhumkar, M. Maheshwari, V.B. Patil and V.B. Pokharkar. Studies on effect of variables by response surface methodology for naproxen microspheres. Indian Drugs, 40: 455-461, 2003.
[16] G.M. Khan. Controlled Release Oral Dosage Forms: Some recent, advances in matrix type drug delivery systems. The Sciences, 1: 350-354, 2001.
[17] D.M. Morkhade, S.V. Fulzele, P.M. Satturwar and S.B. Joshi. Gum copal and gum dammar: novel matrix forming material for sustained drug delivery. Indian J Pharm Sci, 68: 53-58, 2006.
[18] T. Higuchi. Mechanism of rate of sustained-action medication. J Pharm Sci., 52: 1145-1149, 1963.
[19] J. Wang and D.R. Flanagan. General solution for diffusion controlled dissolution of spherical particles.1.theory. J Pharm Sci, 88:731-738, 1999.
[20] S Bolton, Analysis of variance, Pharmaceutical statistics-practical and clinical application. Marcel Dekker, New York, 1997.
Copyright © Priory Lodge Education Limited 2007
First Published June 2007
Click
on these links to visit our Journals:
Psychiatry
On-Line
Dentistry On-Line | Vet
On-Line | Chest Medicine
On-Line
GP
On-Line | Pharmacy
On-Line | Anaesthesia
On-Line | Medicine
On-Line
Family Medical
Practice On-Line
Home • Journals • Search • Rules for Authors • Submit a Paper • Sponsor us
All pages in this site copyright ©Priory Lodge Education Ltd 1994-


