|
|
Vesicular Approach for Drug Delivery into or Across the Skin: Current Status and Future Prospects |
Subheet Jain1*, D. Mishra2, A. Kuksal1, A.K. Tiwary1 and N.K. Jain2
ABSTRACT
The literature is abounding with attempts made repeatedly and sometimes successfully to carry agents into the body through the intact skin by using lipid suspension. The success of systemic drug delivery from liposomal formulation after topical application is low because of the inability of such vesicles to pass through the narrow (< 30 nm) intercellular passage in the outer skin layers. Ethosomes and elastic liposomes also known as Transfersomes¨ are modified lipid carriers that enable drugs to reach into deeper skin layers and/or the systemic circulation. These are soft, malleable vesicles tailored for enhanced systemic delivery of drugs. This article reviews the work carried out in vitro, in vivo in both animal and humans with various ethosomal and transfersomal formulations with particular emphasis on ethosomes. Ethosomes represent a lipid vesicular carrier system embodying ethanol in relatively high concentration and are very efficient in delivering drugs into and across the skin. Unlike classic liposomes, that are known to mainly deliver drugs to outer layers of skin, ethosomes penetrate through the stratum corneum and deliver drugs to the deeper layers of skin.
Key words: vesicular carriers, skin, transdermal, topical, delivery, elastic liposomes, ethosomes.
1. INTRODUCTION
One of the major advances in vesicle research was the finding that some modified vesicles possessed properties that allowed them to successfully deliver drugs in deeper layers of skin. Transdermal delivery is important because it is a noninvasive procedure for drug delivery. Further, problem of drug degradation by digestive enzymes after oral administration and discomfort associated with parenteral drug administration can be avoided. It is the most preferred route for systemic delivery of drugs to pediatric, geriatric and patients having dysphasia. Hence, transdermal dosage forms enjoy being the most patient compliant mode of drug delivery [1, 2].
Despite the promise, there were many problems that researchers had to face with while attempting successful transdermal drug delivery. The skin is a multi-layered structure made up of stratum corneum (SC), the outermost layer, under which lies the epidermis and dermis. Within these layers of skin are interspersed fibroblasts, hair follicles and sweat glands that originate in the dermis blood supply. The almost unsurmountable nature of SC is a major challenge for systemic delivery of percutaneously applied drugs [3]. The Òbrick and mortarÓ arrangement of corneocytes, flattened mononucleated keratinocytes, with interspersed lipids and proteins makes the SC approximately 1000 times less permeable than other biological membranes. Furthermore, it is even more difficult for anything to penetrate to the deeper strata of skin [4, 5].
2. RATIONALE FOR TRANSDERMAL DRUG DELIVERY
Given that the skin offers such an excellent barrier to molecular transport, the rationale for this delivery strategy needs to be carefully identified. There are several instances where the most convenient drug intake methods (the oral route) were not feasible and alternative routes had to be sought. Although, intravenous introduction of the medicament avoids many of these shortfalls (such as gastrointestinal and hepatic metabolism), its invasive and apprehensive nature (particularly for chronic administration) has encouraged the search for alternative strategies. Transdermal drug delivery (TDD) offers several distinct advantages including relatively large and readily accessible surface area (1Ð2 m2) for absorption, ease of application and termination of therapy. Further, evolution of better technologies for delivering drug molecules, safe penetration enhancers and the use of vesicular carriers have rejuvenated the interest for designing TDD system for drugs that were thought to be unfit for transdermal delivery [6-10].
3. VESICULAR APPROACHES FOR TRANSDERMAL/TOPICAL DRUG DELIVERY
Drug encapsulated in lipid vesicles prepared from phospholipids and nonionic surfactants is known to-be transported into and across the skin [11]. Lipids present in the skin contribute to the barrier properties of skin and prevent systemic absorption of drugs. Due to the amphiphilic nature, lipid vesicles may serve as non-toxic penetration enhancer for drugs. In addition, vesicles can be used for encapsulating hydrophilic and lipophilic as well as low and high molecular weight drugs. Therefore, these lipid rich vesicles are hypothesized to carry significant quantity of drugs across the skin thus, enhancing the systemic absorption of drugs.
Drug delivery from liposomes in transdermal formulation has been studied for many purposes but unstable nature and poor skin permeation limits their use for topical delivery [12-19]. In order to increase the stability of liposomes, the concept of proliposomes was proposed [20]. This approach was extended to niosomes, which exhibited superior stability as compared to liposomes [21]. However, due to poor skin permeability, liposomes and niosomes could not be successfully used for systemic drug delivery and their use was limited for topical use [22]. To overcome problems of poor skin permeability Cevc et al. [23] and Touitou et al. [24] recently introduced two new vesicular carrier systems transfersomes and ethosomes, respectively for non-invasive delivery of drugs into or across the skin. Transfersomes¨ and ethosomes incorporated edge activators (surfactants) and penetration enhancers (alcohols and polyols), respectively, to influence the properties of vesicles and stratum corneum [25].
The poor skin permeation characteristic of conventional liposomes became controversial after the introduction of transfersomes. The inventors claimed that tran
sfersomes, being ultradeformable (up to 10 times that of an liposome), could squeeze through pores in stratum corneum (less than one-tenth the vesicleÕs diameter) [26-32]. Thus, sizes up to 200Ð300 nm could penetrate intact skin. Transfersomes require a hydration gradient (non-occluded condition) to be able to penetrate skin [33, 34]. It was proposed that when the skin was not occluded, gradient developed from the (relatively) dry skin surface towards waterlogged viable tissues drives transfersomes through the horny layer. Hence, when the vesicles were applied to non-occluded (dry) skin surface, it resulted in partial dehydration of transfersomes. Furthermore, phospholipids present in these vesicles are known to avoid dry surroundings. Therefore, for vesicles to remain maximally swollen, transfersomes followed the local hydration gradient and penetrated into more strongly hydrated skin layers deeper into the viable epidermis and dermis. On the other hand, conventional liposomes confined themselves to surface or upper layers of stratum corneum, where they dehydrated and fused with skin lipids [35-37].
Remarkable results were claimed for better skin permeation ability of transfersomes. Results indicated that as much as 50% of a topically applied dose of insulin-penetrated skin in vivo in 30 min [25]. Recently, Honeywell-Ngugen et al. [38, 39] observed enhanced delivery of pergolide using elastic liposomes. El-Maghraby et al. [40-43] reported better skin permeation ability of transfersomes containing oestradiol and 5-Fluorouracil as a model drug. A combination of iontophoresis and ultradeformable liposomes was found to enhance the delivery of estradiol [44]. Similarly, transfersomes have been investigated for transcutaneous delivery of dipotassium glycyrrhizinate [45] and cyclosporin A [46]. Hofer et al. [47-49] and Lahmann et al. [50] reported better delivery of immunomodulatory proteins Interleukin-2 and Interferon-a after topical application. Kim et al. [51] prepared ultradeformable cationic liposomes for enhanced transfection efficiency in different cell lines. vanden Bergh et al. [52-55] have proved better skin permeation ability of these elastic liposomes. Paul et al. [56, 57] and Paul and Cevc [58] in different studies reported the use of transfersomes for transdermal immunization. Gupta et al. [59] also recently reported the use of transfersomal formulation for transdermal immunization. Lau et al. [60, 61] suggested the new use of elastic liposomes for the topical treatment of skin cancer. Better systemic delivery of dexamethasone [62-64], norgestrel [65], diclofenac [66, 67] and zidovudine [68] have been observed for transfersomal formulations. A significant (5-10 fold) better skin permeation was observed from transfersomes as compared to that from conventional liposomal formulation. In the light of above reports transfersomes have been suggested to possess better skin permeation ability as compared to conventional liposomes.
4. ETHOSOMES
4.1 Composition
The ethosomes are vesicular carrier comprise of hydroalcoholic or hydro/alcoholic/glycolic phospholipid in which the concentration of alcohols or their combination is relatively high. Typically, ethosomes may contain phospholipids with various chemical structures like phosphatidylcholine (PC), hydrogenated PC, phosphatidic acid (PA), phosphatidylserine (PS), phosphatidylethanolamine (PE), phosphatidylglycerol (PPG), phosphatidylinositol (PI), hydrogenated PC, alcohol (ethanol or isopropyl alcohol), water and propylene glycol (or other glycols) [69]. Such a composition enables delivery of high concentration of active ingredients through skin. Drug delivery can be modulated by altering alcohol: water or alcohol-polyol: water ratio. Some preferred phospholipids are soya phospholipids such as Phospholipon 90 (PL-90). It is usually employed in a range of 0.5-10% w/w. Cholesterol at concentrations ranging between 0.1-1% can also be added to the preparation. Examples of alcohols, which can be used, include ethanol and isopropyl alcohol. Among glycols, propylene glycol and Transcutol are generally used. In addition, non-ionic surfactants (PEG-alkyl ethers) can be combined with the phospholipids in these preparations. Cationic lipids like cocoamide, POE alkyl amines, dodecylamine, cetrimide etc. can be added too. The concentration of alcohol in the final product may range from 20 to 50%. The concentration of the non-aqueous phase (alcohol and glycol combination) may range between 22 to 70% (Table 1) [70].
Table 1. Different Additives Employed In Formulation of Ethosomes
Class |
Example |
Uses |
| Phospholipid |
Soya phosphatidyl choline Egg phosphatidyl choline Dipalmityl phosphatidyl choline Distearyl phosphatidyl choline |
Vesicles forming component |
| Polyglycol |
Propylene glycol Transcutol RTM |
As a skin penetration enhancer
|
| Alcohol |
Ethanol Isopropyl alcohol |
For providing the softness for vesicle membrane As a penetration enhancer |
| Cholesterol |
Cholesterol |
For providing the stability to vesicle membrane |
| Dye |
Rhodamine-123 Rhodamine red Fluorescene Isothiocynate (FITC) 6- Carboxy fluorescence |
For characterization study |
| Vehicle |
Carbopol Ð934 |
As a gel former |
4.2 Influence of high alcohol content
Ethanol is an established efficient permeation enhancer [71, 72] and is present in quite high concentration (20-50%) in ethosomes. However, due to the interdigitation effect of ethanol on lipid bilayers, it was commonly believed that vesicles could not coexist with high concentration of ethanol [73].
Touitou [69] discovered and investigated lipid vesicular systems embodying ethanol in relatively high concentration and named them ethosomes. The basic difference between liposomes and ethosomes lies in their composition. The synergistic effect of combination of relatively high concentration of ethanol (20-50%) in vesicular form in ethosomes was suggested to be the main reason for their better skin permeation ability. The high concentration of ethanol (20-50%) in ethosomal formulation could disturb the skin lipid bilayer organization. Therefore, when integrated into a vesicle membrane, it could give an ability to the vesicles to penetrate the SC. Furthermore, due to high ethanol concentration the ethosomal lipid membrane was packed less tightly than conventional vesicles but possessed equivalent stability. This allowed a softer and malleable structure giving more freedom and stability to its membrane, which could squeeze through small openings created in the disturbed SC lipids [70, 74]. In addition, the vesicular nature of ethosomal formulations could be modified by varying the ratio of components and chemical structure of the phospholipids. The versatility of ethosomes for systemic delivery is evident from the reports of enhanced delivery of quite a few drugs like acyclovir [75], minoxidil [76], triphexyphenidyl [77], testosterone [24], cannabidol [78] and zidovudine [79].
4.3 Method for Preparing Ethosomes
Ethosomal formulation may be prepared by hot or cold method as described below. Both the methods are convenient, do not require any sophisticated equipment and are easy to scale up at industrial level.
4.3.1 Cold Method
This is the most common method utilized for the preparation of ethosomal formulation. In this method phospholipid, drug and other lipid materials are dissolved in ethanol in a covered vessel at room temperature by vigorous stirring with the use of mixer. Propylene glycol or other polyol is added during stirring. This mixture is heated to 300C in a water bath. The water heated to 300C in a separate vessel is added to the mixture, which is then stirred for 5 min in a covered vessel. The vesicle size of ethosomal formulation can be decreased to desire extend using sonication [79] or extrusion [80] method. Finally, the formulation is stored under refrigeration [70].
4.3.2 Hot method
In this method phospholipid is dispersed in water by heating in a water bath at 400C until a colloidal solution is obtained. In a separate vessel ethanol and propylene glycol are mixed and heated to 400C. Once both mixtures reach 400C, the organic phase is added to the aqueous one. The drug is dissolved in water or ethanol depending on its hydrophilic/ hydrophobic properties [69, 70]. The vesicle size of ethosomal formulation can be decreased to the desire extent using probe sonication or extrusion method.
4.4 Physicochemical Characterizations and Properties of Ethosomal Formulation
4.4.1 Vesicle Morphology
Visualization by electron microscopy reveals an ethosomal formulation exhibited vesicular structure 300-400 nm in diameter. The vesicles seem to be malleable as evident by their imperfect round shape (Fig. 1).
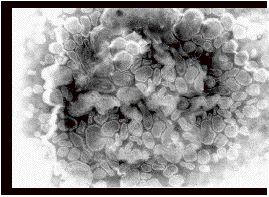
Fig. (1). Morphological characterizations of ethosomal formulation by Transmission Electron Microscopy (TEM). Magnification (x 80, 000)
This characteristic was attributed to the fluidizing effect of ethanol on phospholipid bilayers. Results of 31P-NMR studies showed that PC formed bilayers in the form of closed vesicles up-to 45% ethanol concentration. Phospholipid in ethosome is packed less tightly and the membrane is more permeable to cations as compared to liposomes that are prepared in the absence of ethanol [24]. Thus, in contrast to the accepted view, dispersion of soyabean PC in 45% ethanol results in the formation of closed bilayers vesicles (ethosomes) that retain some barrier properties [81] (Table 2)
Table 2. Methods for the Characterization of Ethosomal Formulation
| Parameters |
Methods |
References |
| Vesicle shape (morphology) |
Transmission electron microscopyScanning electron microscopy |
[125, 126] |
| Entrapment efficiency |
Mini column centrifugation method Fluorescence spectrophotometry |
[127, 128] |
| Vesicle size and size distribution |
Dynamic light scattering method |
[41] |
| Vesicle Skin interaction study |
Confocal laser scanning microscopy Fluorescence microscopy Transmission electron microscopy Eosin-Hematoxylin staining |
[129, 130] |
| Phospholipid-ethanol interaction |
31P NMR Differential scanning calorimeter |
[24, 41]. |
| Degree of deformability |
Extrusion method |
[64, 67] |
| Zeta potential |
Zeta meter |
[77] |
| Turbidity |
Nephalometer |
[77] |
| In vitro drug release study |
Franz diffusion cell with artificial or biological membrane, Dialysis bag diffusion |
[77-79] |
| Drug deposition study |
Franz diffusion cell |
[64, 67] |
| Stability study |
Dynamic light scattering method Transmission electron microscopy |
[24] |
4.4.2 Drug Entrapment Efficiency
Differential scanning calorimetry thermograms and anisotropy measurement of AVPC (a fluorescent analog of phosphatidylcholine), revealed that ethosomes possessed lower Tm compared to classical liposomes and that the bilayers had a high degree of fluidity. This imparted a soft and malleable character to the vesicles. Godin and Touitou [82] used confocal laser scanning microscopy (CLSM) to show that ethosomes can efficiently entrap both hydrophobic and hydrophilic fluorescent probes. Similar results were obtained using ultra-centrifugation method to measure entrapment of different drugs [83]. Efficient loading of both hydrophobic and hydrophilic drugs was confirmed by using hydrophilic 6-carboxyfluorescein and hydrophobic Rhodamine 123 fluorescence markers [79]. The ability of ethosomes to efficiently entrap lipophilic and hydrophilic drugs can be explained by the high degree of lamellarity and by the presence of ethanol in the vesicles. In addition, ethosomal formulations possess greater entrapment capability than liposomes. Dayan and Touitou [77] have shown that entrapment efficiency of trihexyphenidyl hydrochloride increased from 36% for liposomes to 75% for ethosomes.
4.4.3 Vesicle Size and Size distribution
The size of ethosomes ranges between tens of nanometers to microns and is influenced by the composition of the formulation. For example, the ethosomal formulation prepared with 30% ethanol and 2% phospholipids showed an average vesicle size of 161 ± 6.0 nm with a very low polydispersity index (Fig. 2).

Fig. (2). Effect of alcohol concentration on the vesicle size of ethosomal formulation, Mean ± S.D. (n = 3)
In the ethanol concentration range of 10-50%, the size of the vesicles decreased with increasing ethanol concentration. The largest vesicles with 235 ± 8.0 nm size were present in the preparation containing 10% ethanol while the smallest vesicles of 91 ± 5.0 nm size were present in preparation containing 50% ethanol. Similarly, a decrease in the vesicle size (from 214 ± 8.0 nm to 82 ± 3.0 nm) was observed with increase in isopropyl alcohol concentration from 10 to 50%. For comparison, conventional liposomes made from the same phospholipids without alcohol by the film forming method had an average size of 388 ± 14 nm. An eight fold increase in phospholipids concentration from 0.5 to 4%, resulted in significant increase in size of ethosomes from 128 ± 5.0 to 216.± 8.0 nm (Fig. 3) [79].
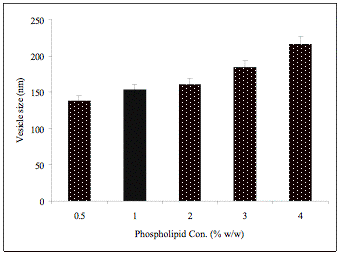
Fig. (3). Effect of phospholipid concentration on the vesicle size of ethosomal formulation, Mean ± S.D. (n = 3)
4.4.4 Permeation Characteristics
One of the most important features of ethosomal formulation is their sustained release characteristic. A significant prolongation of zidovudine release across artificial membrane from ethosomal formulation as compared to drug solution was observed (Fig. 4). The cumulative amount of zidovudine released in 24 hr from ethosomal formulation was 38.4 ± 1.2 % as compared to 92.5 ± 2.1% from the drug solution [79].
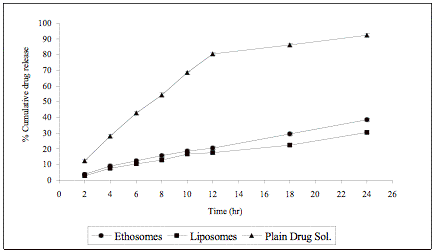
Fig. (4). Comparative cumulative drug release of zidovudine after 24 hr from ethosomal formulation, liposomal formulation and plain drug solution.
In vitro and in vivo skin permeation studies have demonstrated the ability of ethosomal formulation to enhance permeation of both hydrophobic and hydrophilic molecules as compared to conventional liposomes (Fig. 5). Different workers have reported 5-10 fold better skin permeation of drugs formulated in ethosomes as compared to conventional liposomal formulation [84, 85]. The in vitro transdermal flux of zidovudine from ethosomal formulation was observed 78.5±2.5 mg/hr/cm2 across rat skin (Fig. 5).
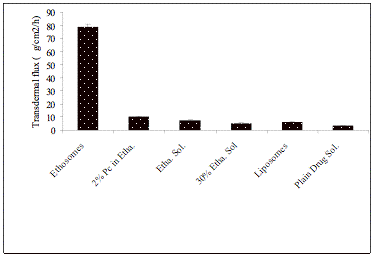
Fig. (5). Transdermal flux of zidovudine through the rat skin from ethosomal formulation and control system. Value represent as mean ± SD (n =3)
This value was eight-fold higher than the flux obtained from formulation containing 2% phospholipids in ethanol (10.2 ± 0.8 mg/h/ cm2), eleven-fold higher than that of ethanolic solution of drug (7.2 ± 0.6 mg/h/cm2), thirteen-fold higher than liposomal formulation (6.1 ± 0.7 mg/h/cm2) and fifteen-fold higher than that of 30 % hydroalcoholic solution of drug (5.2±0.5 mg/h/cm2). A significant difference between permeation of zidovudine from ethosomal formulation and that from ethanolic solution (P > 0.05) indicated that the ethosomes were more effective in transcutaneous delivery.
Ethanol has long been known to have permeation enhancement property. However, the permeation enhancement from ethosomes was much greater than would be expected from ethanol alone, suggesting some kind of synergistic mechanism between ethanol, vesicles and skin lipids. Thus, ethanol that was earlier considered harmful to conventional liposomal formulations, provided flexible characteristics to ethosomes, which allows them to easily penetrate into deeper layers of the skin. In addition, the contribution of interaction between phospholipid vesicles with stratum corneum as proposed by Kirajavainen et al. [86] in enhancing the permeability of skin cannot be neglected.
4.4.5 Vesicle Skin Interaction Study
For evaluating the mechanism of better skin permeation of ethosomal formulation different visualization techniques e.g. transmission electron microscopy, eosin-hematoxylin staining, fluorescence microscopy and confocal scanning laser microscopy (CSLM) have been used. Often, when used in combination these visualization techniques gave better idea about structure modulation and penetration pathways of vesicles [87-92].
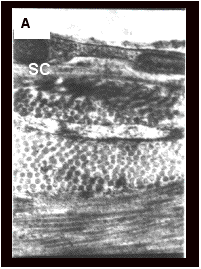
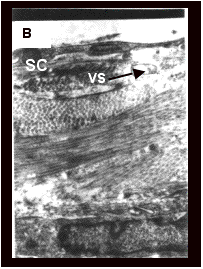
![]()

Fig. 6.A-C represents the transmission
electron micrographs of phosphate buffer saline (PBS), conventional liposomal
and ethosomal formulation treated rat skin. After treatment with ethosomal
formulation, areas of lipids with electron dense material were visualized
deeper down in the stratum corneum that is fixed only by osmium tetraoxide
(OsO4). Since, SC lipids lamellae cannot be fixed by OsO4
[87, 93], it was suggested that the dense material originated from the vesicles.
These OsO4 fixed lipid areas containing electron dense material
were not observed in PBS and conventional liposomal formulation treated skin
(Fig. 6A-B). 
Fig. (7A-D). Fluorescence photomicrograph of rat skin after application of hydrophilic fluorescence probe 6-carboxyfluorescein from (A) Liposomal formulation (x 100); (B) Ethosomal formulation (x100) and Rhodamine 123 from (C) Liposomal formulation; (x 100) (D) Ethosomal formulation (x100). SC = Stratum corneum, E = Epidermis, D = Dermis; FL = Fibrous layer, Ad = Adipose tissue, Ve = Vesicular stacks
No ultrastructural changes were observed in cell layers below the stratum corneum indicating that rigid liposomal formulation did not induce any changes in the ultrastructure of stratum corneum and accumulated only in the top layer of the skin. These results illustrated that liquid state vesicles might act not only in superficial stratum corneum layers, but may also induce liquid perturbations in deeper layers of the SC, while gel state vesicles interacted only with the outermost layers in the SC. This might explain the difference in drug permeation enhancement between ethosomal and conventional liposomal formulation. In addition, fusion of conventional liposomal vesicles on top of the stratum corneum might also act as additional barrier for diffusion of drugs and therefore inhibit skin permeation.
To support the result of TEM study Jain et al. [79] performed histological studies in order to visualize the changes in the ultrastructure of stratum corneum. The results of eosin-haematoxyline staining study showed that ethosomal formulation affected the ultrastructure of stratum corneum. No change in the ultrastructure of viable tissue (epidermis or dermis) could be observed after treatment with conventional liposomal formulation [94].
Fluorescence photomicrographs of the skin after a 6 hr application of Rhodamine 123 (lipophilic probe) or 6-CF (hydrophilic probe) loaded liposomal and ethosomal formulation are shown in Fig. 7A-D.
Penetration from conventional liposomes was only to upper layer of skin (stratum corneum). Deep penetration from alcohol free liposomes was almost negligible (Fig. 7A, C). In contrast enhanced delivery of 6-CF and Rhodamine 123 in terms of depth and quantity (dermis layer) was observed using the ethosomal carrier (Fig. 7B, D). These results supported the results of skin permeation studies and showed the feasibility of using ethosomal formulation for delivering drugs into the deeper layers of skin or across the skin. [79].
Touitou et al. [24] reported the ability of ethosomes to deliver lipophilic molecules to deep layers of skin using a lipophilic fluorescent probe, Rhodamine red (RR) by CSLM. They found that intensity of fluorescence was much greater when ethosomal system was applied as compared to that when either a hydroalcoholic solution containing the same concentration of ethanol or an alcohol free liposomal system was applied. RR contained in ethosomes penetrated the mouse skin to a depth of approximately 140 mm. The probe fluorescence intensity was significantly greater from the ethosomal preparation whereas, deep penetration from conventional liposomal formulation was almost negligible. Similarly, Godin and Touitou [82] reported better skin permeation of fluoreceine isothiocyanate-bacitracin ethosomal formulation to deeper layer of skin as determined by CLSM (Table 3).
Table 3. Vesicle Skin Interaction with Rat Skin
| S.No. |
Formulation |
Physical state |
Adsorption |
Structural changes |
|
| Intercellular |
Intracellular |
||||
| 1 |
Rigid Liposomes |
Gel |
+ |
- |
- |
| 2 |
Ethosomal formulation |
Liquid |
- |
+ |
+ |
| 3 |
Plain drug |
Liquid |
ND |
- |
- |
Summary of the interaction between the different formulations and the stratum corneum.
+ = Frequently observed - = Not observed ND= Not determined
4.5 Proposed Mechanism of Skin Permeation of Ethosomes
Fig. 8 showed the schematic representation of mechanism of skin permeation of ethosomes. The stratum corneum lipid multilayers at physiological temperature are densely packed and highly conformationally ordered. Ethosomal formulations contain ethanol in their composition that interacts with lipid molecules in the polar headgroup regions, resulting in an increased fluidity of the SC lipids. The high alcohol content is also expected to partial extract the SC lipids. These processes are responsible for increasing inter and intracellular permeability of ethosomes. In addition, ethanol imparts flexibility to the ethosomal membrane that shall facilitate their skin permeation. The interdigitated, malleable ethosome vesicles can forge paths in the disordered SC and finally release drug in the deep layers of skin. The transdermal absorption of drugs could then result from fusion of ethosomes with skin lipids. This is expected to result in drug release at various points along the penetration pathway [95-97].
4.6 Different Studies Related to the Application of Ethosomes as a Carrier System
Various studies employing ethosomal formulation
have shown better skin permeability of drugs. The uses of ethosomes as carrier
system for transdermal/topical drug delivery are summarized below (Table 4).
Table 4. Application of Ethosomes as a Drug Carrier
| Drug |
Results |
References |
| NSAIDS (Diclofenac) |
¯ Selective delivery of drug to desired side for prolong period of time |
[69, 70] |
| Acyclovir
|
¯ Increase skin permeation ¯ Improved in biological activity two to three times ¯ Improved in Pharmacodynamic profile |
[75]
|
| Insulin |
¯ Significant decrease in blood glucose level ¯ Provide control release |
[120] |
| Trihexyphenidyl hydrochloride |
¯ Improved transdermal flux ¯ Provide controlled release ¯ Improved patient compliance ¯ Biologically active at dose several times lower than the currently used formulation |
[77] |
DNA |
¯ Better expression of genes ¯ Selective targeting to dermal cells |
[104] |
| Antibiotic Cannabidol Erythromycin |
¯ Improved skin deposition ¯ Improved biological activity ¯ Prolonging drug action |
[78]
|
Bacitracin |
¯ Improved dermal deposition ¯ Improved intracellular delivery ¯ Increased bioavailability |
[82] |
| Anti-HIV agents Zidovudine Lamivudine |
¯ Improved transdermal flux ¯ Improved in biological activity two to three times ¯ Prolonging drug action ¯ Reduced drug toxicity ¯ Affected the normal histology of skin |
[79]
|
| Azelaic acid |
¯ Prolong drug release |
[85] |
| Ammonium glycyrrhizinate |
¯ Improved dermal deposition exhibiting sustained release ¯ Improved biological anti-inflammatory activity |
[123] |
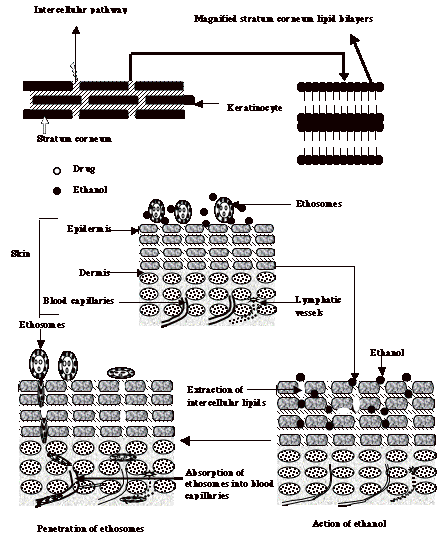
Fig. (8). Proposed mechanism for penetration of molecule from ethosomal system across the lipid domain of stratum corneum
4.6.1 Pilosebaceous Targeting
Hair follicles and sebaceous glands are increasingly being recognized as potentially significant elements in the percutaneous drug delivery. Interest in pilosebaceous units has been directed towards their use as depots for localized therapy, particularly for the treatment of follicle-related disorders such as acne or alopecia. Furthermore, considerable attention has also been focused on exploiting the follicles as transport shunts for systemic drug delivery [98]. With the purpose of pilosebaceous targeting, Maiden et al. [99] prepared and evaluated minoxidil ethosomal formulation. Minoxidil is a lipid-soluble drug used topically on the scalp for the treatment of baldness. Conventional topical formulation has very poor skin permeation and retention properties. It was found that the quantity of minoxidil accumulated into nude mice skin after application of its ethosomal formulation was 2.0, 7.0 and 5.0 fold higher as compared to ethanolic phospholipids dispersion, hydroetanolic solution and ethanolic solution of drug each containing 0.5% of the drug. These results showed the possibility of using ethosomes for pilosebaceous targeting of minoxidil to achieve its better clinical efficacy.
4.6.2 Transdermal Delivery of Hormones
Oral administration of hormones is associated with problems like high first pass metabolism, low oral bioavailability and several dose dependent side effects. In addition, along with these side effects oral hormonal preparations relying highly on patient compliance. The risk of failure of treatment is known to increase with each pill missed [100].
Touitou et al. [24] compared the skin permeation potential of testosterone ethosomes (Testosome) across rabbit pinna skin with marketed transdermal patch of testosterone (Testoderm¨ patch, Alza). They observed nearly 30-times higher skin permeation of testosterone from ethosomal formulation as compared to that marketed formulation. The amount of drug deposited was significantly (p <0.05) higher in case of ethosomal formulation (130.76 ± 18.14 and 18.32 ± 4.05 mg at the end of 7 hr for Testosome and Testoderm¨, respectively). The AUC and Cmax of testosterone significantly improved after application of Testosome as compared to Testoderm. Hence, both in vitro and in vivo studies demonstrated improved skin permeation and bioavailability of testosterone from ethosomal formulation. This group in their further study designs the testosterone nonpatch formulation to reduce the area of application [101]. They have found that with ethosomal testosterone formulation area of application required to produce the effective plasma concentration was 10 times less than required by commercially gel (AndroGel¨) formulation.
4.6.3 Delivery of anti-parkinsonism agent
Dayan and Touitou [77] prepared ethosomal formulation of psychoactive drug trihexyphenidyl hydrochloride (THP) and compared its delivery with that from classical liposomal formulation. THP is a M1 muscarinic receptors antagonist and used in the treatment of Parkinson disease. THP has a short biological half-life (3hr) and its oral administration is difficult due to motor disorders and neurogical manifestations associated with parkinsonian syndrome [103]. THP ethosomal formulation when visualized under transmission and scanning electron microscope found to consist of small, phospholipid vesicles. The value of transdermal flux of THP through nude mouse skin from ethosomes was 87, 51 and 4.5-times higher than that from liposome, phosphate buffer and hydroethanolic solution, respectively. The quantity of THP remaining in skin at the end of 18 hr was significantly higher after application of ethosomes than after application of liposome or hydroethanolic solution (control). These results indicated better skin permeation potential of ethosomal-THP formulation and its use for better management of Parkinson disease.
4.6.4 Transcellular Delivery
Touitou et al. [84] investigated the efficiency of transcellular delivery of ethosomes in Swiss albino mice 3T3 fibroblast. The probes chosen for study were D-289 [4-(4-(diethylamino) styryl-N-methylpyridinum iodide], rhodamine red [dihexadecanoylglycerophosphoethanolamine] and fluorescent phosphatidylcholine. The penetration of these fluorescent probes into fibroblasts and nude mice skin was examined by CLSM (Confocal Laser Scanning Microscopy) and FACS (Fluorescent Activated Cell Sorting) techniques. CLSM micrograph showed that significant quantity of probe was penetrated into the cells when incorporated into ethosomes as evident from the high intensity of fluorescence. In comparison, incorporation into hydroethanolic solution or classic liposomes produced almost negligible fluorescence. The intracellular presence of each of the three probes tested was evident after 3 min. of incubation. Enhanced delivery of the hydrophilic calcein and lipophilic rhodamine red (RR) probe to nude mice skin was also observed when incorporated into ethosomes. Calcein penetrated the skin to a depth of 160, 80 and 60 mM from ethosomes, hydroethanolic solution and liposomes, respectively. Maximum fluorescence intensities measured for RR delivered from ethosomes, hydroethanolic solution and liposomes were 150, 40 and 20 arbitrary units (AU), respectively. Fibroblasts viability tests showed that the ethosomal carrier was not toxic to the cultured cells [84].
Touitou et al. in their further study demonstrated better intracellular uptake of bacitracin [82], DNA [104] and erythromycin [105] using CLSM and FACS techniques in different cell lines. Better cellular uptake of anti-HIV drug zidovudine and lamivudine in MT-2 cell line from ethosomes as compared to the marketed formulation suggested ethosomes to be an attractive clinical alternative for anti-HIV therapy [106].
4.5.5 Topical Delivery of DNA
Many environmental pathogens attempt to enter the body through the skin. Skin therefore, has evolved into an excellent protective barrier, which is also immunologically active and able to express the gene [107]. On the basis of above facts another important application of ethosomes is to use them for topical delivery of DNA molecules to express genes in skin cells [108]. Touitou et al. [109] in their study encapsulated the GFP-CMV-driven transfecting construct into ethosomal formulation. They applied this formulation to the dorsal skin of 5-week male CD-1 nude mice for 48 hr. After 48 hr, treated skin was removed and penetration of green fluorescent protein (GFP) formulation was observed by CLSM. It was observed that topically applied ethosomes-GFP-CMV-driven transfecting construct enabled efficient delivery and expression of genes in skin cells. It was suggested that ethosomes could be used as carriers for gene therapy applications that require transient expression of genes. These results also showed the possibility of using ethosomes for effective transdermal immunization. Gupta et al. [59] recently reported immunization potential using transfersomal formulation. Hence, better skin permeation ability of ethosomes opens the possibility of using these dosage forms for delivery of immunizing agents
4.6.6 Delivery of Anti-Arthritis Drug
Topical delivery of anti-arthritis drug is a better option for its site-specific delivery and overcomes the problem associated with conventional oral therapy. Cannabidol (CBD) is a recently developed drug candidate for treating rheumatoid arthritis. Its oral administration is associated with a number of problems like low bioavailability, first pass metabolism and GIT degradation [110]. To overcome the above mention problem Lodzki et al. [78] prepared CBD-ethosomal formulation for transdermal delivery. Results of the skin deposition study showed significant accumulation of CBD in skin and underlying muscles after application of CBD-ethosomal formulation to the abdomen of ICR mice Plasma concentration study showed that steady state level was reached in 24 hr and maintained through 72 hr. Significantly increased in biological anti-inflammatory activity of CBD-ethosomal formulation was observed when tested by carrageenan induced rat paw edema model. Finally, it was concluded that encapsulation of CBD in ethosomes significantly increased its skin permeation, accumulation and hence its biological activity.
4.6.7 Delivery of Antibiotics
Topical delivery of antibiotics is a better choice for increasing the therapeutic efficacy of these agents. Conventional oral therapy causes several allergic reactions along with several side effects. Conventional external preparations possess low permeability to deep skin layers and subdermal tissues [111]. Ethosomes can circumvent this problem by delivering sufficient quantity of antibiotic into deeper layers of skin. Ethosomes penetrate rapidly through the epidermis and bring appreciable amount of drugs into the deeper layer of skin and suppress infection at their root. With this purpose in mind Godin and Touitou [82, 105] prepared bacitracin and erythromycin loaded ethosomal formulation for dermal and intracellular delivery. CLSM experiments revealed that ethosomes facilitated the co-penetration of antibiotic and phospholipid into cultured 3T3 Swiss albino mice fibroblasts. The data obtained by CLSM experiment was confirmed by FACS techniques and it was found that ethosomes penetrated the cellular membrane and released the entrapped drug molecules within the cells. The results of this study showed that the ethosomal formulation of antibiotic could be highly efficient and would over come the problems associated with conventional therapy.
4.5.8 Delivery of Anti-Viral Drugs
Zidovudine is a potent antiviral agent acting on acquired immunodeficiency virus. Oral administration of zidovudine is associated with strong side effects. Therefore, an adequate zero order delivery of zidovudine is desired to maintain expected anti-AIDS effect [112, 113]. In a recent study the optimized ethosomal formulation exhibited a transdermal flux of 78.5±2.5 mg/cm2/h across rat skin, while the hydroethanolic solution gave a flux of only 5.2±0.5 mg/cm2/h of zidovudine. The flux from ethanolic solution was found to be 7.2±0.6 mg/cm2/h. Jain et al. [79] concluded from this study that ethosomes could increase the transdermal flux, prolong the release and present an attractive route for sustained delivery of zidovudine.
Acyclovir is another anti-viral drug that widely used topically for treatment of Herpes labialis [114-115]. The conventional marketed acyclovir external formulation is associated with poor skin penetration of hydrophilic acyclovir to dermal layer resulting in weak therapeutic efficiency [116]. It is reported that the replication of virus takes place at the basal dermis [117-119]. To overcome the problem associated with conventional topical preparation of acyclovir, Horwitz et al. [75] formulated the acyclovir ethosomal formulation for dermal delivery. They have clinically evaluated its performance in a double blind, randomized study with marketed formulation of acyclovir (Zovirax, Glaxo-Wellcome) in terms of time to crust formation, time to loss of crust and proportions of lesions not progressive beyond the popular stage (abortive lesions). Significant improvement in all evaluated clinical parameters was observed when disorder was treated with ethosomal formulation in comparison to marketed formulation. The average time to crusting of lesions was 1.6 vs 4.3 days in the parallel arm and 1.8 vs. 3.5 days in the crossover arm (P<0.025) for ethosomal acyclovir and Zovirax, respectively. Hence, shorter healing time and higher percentage of abortive lesions were observed when acyclovir was loaded into ethosomes.
4.6.9 Delivery of Problematic drug molecules
The oral delivery of large biogenic molecules such as peptides or proteins is difficult because they are completely degraded in the GI tract. Non-invasive delivery of proteins is a better option for overcoming the problems associated with oral delivery [120-121]. Dkeidek and Touitou [122] investigated the effect of ethosomal insulin delivery in lowering blood glucose levels (BGL) in vivo in normal and diabetic SDI rats. In this study a Hill Top patch containing insulin ethosomes was applied on the abdominal area of an overnight fated rat. The result showed that insulin delivered from this patch produced a significant decrease (up to 60%) in BGL in both normal and diabetic rats. On the other hand, insulin application from a control formulation was not able to reduce the BGL.
Verma and Fahr [80] reported the cyclosporin A ethosomal formulation for the treatment of inflammatory skin disease like psoriasis, atopic dermatitis and disease of hair follicle like alopecia areata etc. They have combined the ethanol with a commercially lipid mixture NAT 8539 contained phosphatidylcholine (73-75%), lyso-phosphatidylcholine (upto 6%), Cephaline (upto 4%) and phosphatidic acid (upto 6%) and natural oils. They have found that cyclosporine vesicles prepared with NAT 8539/ethanol (10/3.3) showed 2.1 fold, NAT 8539/ethanol (10/10) showed a 4.4 fold and NAT 8539/ethanol (10/20) showed a 2.2 higher deposition of cyclosporine into SC as compared to vesicle made of NAT 8539 without ethanol. The result of skin deposition study was confirmed by CLSM study. The result obtained was similar to skin deposition study. As the concentration of ethanol increased the depth and intensity of fluorescence was increased. Formulation NAT 8539/ethanol ((10/10) produced a fairly homogeneous bright fluorescence throughout the SC. They have concluded that ethanolic liposomal formulation can be used for the topical delivery of problematic drug molecules like cyclosporine whose oral delivery is difficult.
Paolino et al. [123] investigated the potential application of ethosomes for dermal delivery of ammonium glycyrrhizinate. Ammonium glycyrrhizinate is naturally occurring triterpenes obtained from Glycyrrhizinate Glabra and useful for the treatment of various inflammatory based skin diseases [124]. In vitro skin permeation experiments showed the significantly (P<0.001) higher cumulative amount of drug permeated from ethosomes (63.2%) than hydroalcoholic solution (22.3%) and aqueous solution (8.9%) of ammonium glycyrrhizinate. They have also evaluated the human skin tolerability using Reflectance Spectrophotometry that is a non-invasive technique to evaluate the carrier toxicity. Ethosomal formulation showed a very good skin tolerability in human volunteer even applied for 48 hr. Biological anti-edema activity also showed the significant enhanced in case of ethosomal formulation as compared to ethanolic or aqueous solution of drug.
5. Future Prospects
Introduction of ethosomes has initiated a new area in vesicular research for transdermal drug delivery. Different reports show a promising future of ethosomes in making transdermal delivery of various agents more effective. Further, research in this area will allow better control over drug release in vivo, allowing physician to make the therapy more effective. Ethosomes offers a good opportunity for the non-invasive delivery of small, medium and large sized drug molecules. The results of the first clinical study of acyclovir-ethosomal formulation support this conclusion. Multiliter quantities of ethosomal formulation can be prepared very easily. It, therefore, should be not before long that the corresponding drug formulation would have found their way into clinics to be tested for widespread usage. Thus, it can be a logical conclusion that ethosomal formulations possess promising future in effective dermal/transdermal delivery of bioactive agents.
| Click on these buttons to visit our journals | ||||||||
| Psychiatry On-Line | Dentistry On-Line | Vet On-Line | Chest
Medicine On-Line | GP On-Line | Pharmacy On-Line | Anaesthesia On-Line | Medicine On-Line | Family
Medical Practice On-Line |
All pages copyright ©Priory Lodge Education Ltd 1994-2006.
REFERENCES
[1] Albery, W.J. and Hadgraft, J. (1979) J. Pharm. Pharmacol. 31,129-139.
[2] Kanitakis, J. (2002) Eur. J. Dermatol. 12, 390-399.
[3] Barry, B.W. (2001) Eur. J. Pharm. Sci. 14, 101-114.
[4] Hadgraft, J. (2001) Int. J. Pharm., 2001, 224, 1-18.
[5] Hadgraft, J. (2001) Skin Pharmacol. Appl. Skin. Physiol. 14, 72-81.
[6] Guy, R.H. and Hadgraft, (1985) J. Pharm. Int., 6, 112-116.
[7] Flynn, G.L. (1990) In Principles of route to route extrapolation for risk assessment, Gerrity, T.R.; Henry, C.J. Ed.; Elsevier Science : Amsterdam, pp 93-127.
[8] Le, V.H. and Lippold, B.C. (1995) Int. J. Pharm. 124, 285-292.
[9] Coderch, L.; de Para, M.; Perex-Cullell, N.; Estelrich, J.; de la Maza, A.; Parra, J.L. (1999) Skin Pharmacology and Applied Skin Physiology. 12, 235-246.
[10] Panchagnula, R.; Pillai, O.; Nair, V.B.; Ramarao, P. (2000) Curr. Opin. Chem. Biol. 4, 468-473.
[11] Schreier, H. and Bouwstra, J. (1994) J. Control. Release. 30, 1-15.
[12] Mezei, M. and Gulusekharam, V. (1980) Life Sci, 26, 1473-1477.
[13] Mezei, M. and Gulusekharam, V. (1982) J Pharm. Pharmacol. 34, 473-474.
[14] Egbaria, K. and Weiner, N. (1990) Adv. Drug. Deliv. Rev. 5, 287-300.
[15] Touitou, E.; Junginger, H.E.; Weiner, N.D.; Nagai, T.; Mezei, M. (1994) J. Pharm. Sci. 83, 1189-1203.
[16] Touitou, E.; Levi-Schaffer, F.; Dayan, N.; Alhaique, F.; Riccieri, F. (1994) Int. J. Pharm. 103, 131-136.
[17] Schmid, M.H. and Korting, H.C. (1994) Crit. Rev. Ther. Drug Carrier Systems. 11, 97-118.
[18] Weiner, N. (1998) Int. J. Pharm. 162, 29-38.
[19] Yarosh, D.B. (2001) Photoimmunology and Photomedicine. 17, 203-212.
[20] Deo, M.R.; Sant, V.P.; Prakash, S.R.; Khopade, A.J.; Banakar, U.V. (1997) J. Biomaterials appli. 12, 77-85.
[21] Vora, B.; Khopade, A.J.; Jain, N.K. (1998) J. Control. Release. 54, 149-165.
[22] Lasch, J.; Laub, P.; Wohlrab, W. (1991) J. Control. Release, 18, 55-58
[23] Cevc, G.; Blume, G.; Schatzlein, A.; Gebauer, D.; Paul, A. (1996) Adv. Drug. Deliv. Rev. 18, 349-378.
[24] Touitou, E.; Dayan, N.; Bergelson, L.; Godin, B.; Eliaz, M. (2000) J. Control. Release. 65, 403-418.
[25] Cevc, G.; Blume, G.; Schatzlein, A. (1997) J. Control. Release. 45, 211-226.
[26] Cevc, G. and Blume, G. (1992) Biochim. Biophys Acta, 1104, 226-232
[27] Cevc, G. (1996) Crit. Rev. Ther. Drug Carrier Syst. 13, 257-388
[28] Fesq, H.; Glockner, A.; Abeck, D.; Ring, J.; Lehmann, J.; Rother, M.; Cevc, G. (2000) J. Invest. Dermatol. 115, 590-590.
[29] Cevc, G. and Blume, G. (2001) Biochim. Biophysic. Acta. 1514, 191-205.
[30] Cevc, G. and Gebauer, D. (2003) Biophys. J. 84, 1010-1024.
[31] Cevc, G. (2003) Clin Pharmacokinet. 42 (5), 461-474.
[32] Cevc, G.; Schþtzlein, A.G.; Richardsen, H.; Vierl, U. (2003) Langmuir, 19, 10753-10763.
[33] Warner, R.R.; Myers, M.C.; Taylor, D.A. (1990) J. Invest. Dermatol. 90, 218.
[34] Gompper, R. and Kroll, D.M. (1995) Phys. Rev. E-52, 4198-5105.
[35] Cevc, G. (2004) Adv. Drug. Deliv. Rev. 56, 675-711.
[36] Cevc, G.; Gebauer, D.; Schatzlein, A.; Blume, G. (1998) Biochim. Biophys. Acta. 1368, 210-225.
[37] Cevc, G.; Schatzlein, A.; Richardsen, H. (2002) Biochim. Biophys. Acta. 1564, 21-30.
[38] Honeywell-Nguyen, P.L.; Frederik, P.M.; Bomans P.H.H.; Junginger, H.E. Bouwstra, J.A. (2002) Pharm. Res. 19(7), 991-997.
[39] Honeywell-Nguyen, P.L.; Frederik, P.M.; Bomans P.H.H.; Junginger, H.E. Bouwstra, J.A. (2003) J. Control. Release. 86, 145-156.
[40] El. Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. (1999) J. Pharm. Pharmacol. 51, 1123-1134.
[41] El. Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. (2000) Int. J. Pharm. 196, 63-74.
[42] El. Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. (2001) J. Pharm. Pharmacol. 53, 1311-1322.
[43] El. Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. (2001) J. Pharm. Pharmacol. 53, 1069-1076.
[44] Essa, E.A.; Bonner, M.C.; Barry, B.W. (2002) Int. J. Pharm. 240, 55-66.
[45] Trotta, M.; Peira, E.; Debernadi, F.; Gallarate, M. (2002) Int. J. Pharm. 241, 319-327.
[46] Guo, J.; Ping, G.; Sun, G.; Jiao, C. (2000) Int. J. Pharm. 194, 201-207.
[47] Hofer, C.; Goble, R.; Deering, P.; Lehmere, A. Breut, J. (1999) Anticancer Res. 19, 1501-1505.
[48] Hofer, C.; Hartung, R.; Gobel, R.; Deering, P.; Lehmer, A. and Breul, J. (2000) World J. Surg. 24, 1187-1189.
[49] Hofer, C.; Randenborge, H.; Lehmer, A.; Hartung, R.; Breul, J. (2004) Cytokine. 78, 112-118.
[50] Lehmann, J.; Theiling, K.; Klede, M.; Ikoma, A.; Schmelz, M.; Rother, M.; von Wossow, P. (2000) Int. J. Immunotherapy. 18, 21Ð26.
[51] Kim, A.; Lee, E.H.; Choi, S.H.; Kim, C.K. (2004) Biomaterials, 25, 305-313.
[52] vanden Bergh, B.A.I.; Salomons, B.I.; Bouwstra, J.A. (1998) Int. J. Pharm. 167, 57-67.
[53] vanden Bergh, B.A.I.; Bouwstra, J.A.; Junginger, H.E.; Wertz, P.W. (19990 J. Control. Release. 62, 367-379.
[54] vanden Bergh, B.A.I.; Vroom, J.; Gerritsen, H.; Junginger, H.E.; Bouwstra, J.A. (1999) Biochim. Biophys Acta. 1461, 155-173.
[55] vanden Bergh, B.A.I.; Wertz, P.W.; Junginger, H.E.; Bouwstra, J.A. (2001) Int. J. Pharm. 217, 13-24.
[56] Paul, A.; Cevc, G.; Bachhawat, B.K. (1995) Eur. J. Immunol. 25, 3521-3524.
[57] Paul, A.; Cevc, G.; Bachhawat, B.K. (1998) Vaccine. 16(2/3), 188-195.
[58] Paul, A. and Cevc, G. (1995) Vaccine Res. 4, 145-150.
[59] Gupta, P.N.; Mishra, V.; Rawat, A.; Jain, S.; Dubey, P K ; Mahor, S.; Chatterji, D.P.; Vyas, S P. (2005) Int. J. Pharm. 293, 73-82.
[60] Lau, K.G., Chopra, S., Maitani, Y., Entrapment of bleomycin in ultra-deformable liposomes. (2003) S. T. P. Pharm. Sci. 13, 237Ð239.
[61] Lau, K.G., Hattori, Y., Chopra, S., Toole, E.A.O., Storey, A., Nagai, T., Maitani, Y. Ultradeformable liposomes containing bleomycin: In vitro stability and toxicity on human cutaneous keratinocyte cell lines. (2005) Int. J. Pharm. 300, 4Ð12.
[62] Jain, S.; Jain, S.; Jain, N.K. (2001) Transfersomes for enhanced delivery of dexamethasone, Proceeding of 28th Conference of CRS, USA, 5207.
[63] Jain, S.; Jain, S.; Jain, N.K. (2002) Drug Delivery Tech. 2(3), 70-71.
[64] Jain, S.; Jain, P.; Jain, N.K. (2003) Drug Dev. Ind. Pharm. 29(90), 1013-1026.
[65] Jain, S.; Sapre, R.; Umamaheshwari, R.B.; Jain, N.K. (2003) Ind. J. Pharm. Sci., 65(2), 152-161.
[66] Jain, S.; Jain, N.; Bhadra, D.; Tiwary, A.K. Jain, N.K. (2002) Sustained and targeted delivery of an analgesic agent using ultradeformable lipid vesicles. Proceeding of the 54rd Indian Pharmaceutical conference, Pune (India), A-67
[67] Jain, S.; Jain, N.; Bhadra, D.; Tiwary, A.K. Jain, N.K. (2005) Current Drug Delivery 2(3), 222-233
[68] Jain, S.; Tiwary, A.K. Jain, N.K. Sustained and targeted transdermal delivery of an anti-HIV agent using elastic liposomal formulation: Mechanism of action (2006) Current Drug Delivery 3(2), 157-164
[69] Touitou, E. Composition of applying active substance to or through the skin., US patent, 5,716,638, 1996.
[70] Touitou, E. Composition of applying active substance to or through the skin., US patent, 5,540,934, 1998.
[71] Braun-Falco, O.; Kortung, H.C.; Maibach, H.I. Liposome Dermatitis, Springer-Verlag, Berlin Heideberg, 1992.
[72] Berner, B.; Liu, P. (1995) Alcohol, In Percutaneous Enhancer, Smith, E.W.; Maibach, H.I., Ed.; CRC Press, Boca Raton, FI, pp 45-60.
[73] Riaz, M.; Weiner, N.; Martin, F. (1998) In Pharmaceutical Dosage forms, Disperse Systems, Liberman, H.A.; Reiger, M.M.; Banker, G.S., Ed.; Marcel Dekker, New-York, Basel, Vol. 2.
[74] Barry, B.W. (2001) Eur. J. Pharm. Sci. 14, 101-114.
[75] Horwitz, E.; Pisanty, S.; Czerninsky R.; Helser, M., Eliav, E., Touitou, E. (1999) Oral Surg Oral Pathol Oral Radiol Endod, 88, 700-05.
[76] Godin, B.; Alkabes, M.; Touitou, E. (1999) Acta Technologiae et Legis Medicament. 10, 107.
[77] Dayan, N. and Touitou, E. (2000) Biomaterials. 21, 1879-1885.
[78] Lodzki, M.; Godin, B.; Rakou, L.; Mechoulam, R.; Gallily, R.; Touitou, E. (2003) J. Control. Release. 93, 377-387.
[79] Jain, S.; Umamaheshwari, R.B.; Bhadra, D.; Jain, N.K. (2004) Ind. J. Pharm. Sci. 66(1), 72-81.
[80] Verma, D.D. and Fahr, A. (2004) Synergistic penetration effect of ethanol and phospholipids on the topical delivery of Cyclosporin A. J. Control Release. 97, 55-66.
[81] Touitou, E.; Dayan, N.; Bergelson L.; Levi-Schaffer F.; Piliponsky, (1998) J. Liposome Res. 8,112-114.
[82] Godin, B. and Touitou, E. (2004) J. Control Release. 187, 1-15
[83] Touitou E. Godin B.; Bujanover S.; Becker Y. (2002) J. Control. Release. 80, 1-7.
[84] Touitou, E.; Godin, B.; Dayan, N.; Weiss, C.; Piliponsky, A.; Levi-Schaffer, F. (2001) Biomaterials. 22, 3053-3059.
[85] Esposito, E.; Menegatti, E.; Cortesi, R. (2004) J Cosmet. Sci. 55(3), 253-64.
[86] Kirajavainen, M.; Urtti, A.; Valjakka-Koskela, R.; Kiesvaara, J.; Monkkonen, J. (1999) Eur. J. Pharm. Sci. 7, 279-286.
[87] vanden Berge, B.A.I.; Swartzendruber, V.A.B.; Geest, J. (1997) J. Microsc., 187, 125-133.
[88] Van Kuijk-Meuwissen, Marly E.M.J.; Junginger, H.E.; Bouwstra, J.A. (1998) Biochim. Biophys. Acta. 1371, 31-39.
[89] Betz, G.; Imboden, R.; Georgios, I. (2001) Int. J. Pharm. 229, 117-129.
[90] Ogiso, T.; Yamaguchi, T.; Iwaki, M.; Tanino T.; Miyake. Y. (2001) J. Drug Targeting. 9(1), 49-53
[91] Grams, Y.Y. and Bouwstra, J.A. (2002) J. Control Release. 83, 253-262.
[92] Bouwstra, J.A.; Honeywell-Nguyen, P.L.; Gooris, G.S.; Ponec, M. (2003) Progress in Lipid Research. 42, 1-36.
[93] Bouwstra, J.A. and Honeywell-Nguyen, P.L. Adv. Drug Deliv. Rev., 2002, 54(1), S41-S55.
[94] Gartner, L.P.; Hiatt, J.L. Color Textbook of Histology, Philadelphia: W.B. Saunders Company, Second Edition, 2001.
[95] Touitou, E.; Alkabes, M.; Dayan, N.; Eliaz, M. (1997) Pharm. Res. 14, S305-S306.
[96] Touitou, E.; Dayan, N.; Bergelson L.; Levi-Scaffer. F.; Pilponsky, A. (1998) J. Lipid Res. 8, 113-114
[97] Godin, B. and Touitou, E. (2003) Crit. Rev. Therp.Drug Carrier Sys, 20(1), 63-102.
[98] Lauer, A.C. Ramachandran, C.; Lieb, L.M.; Niemiec, S. Weiner, N.D. (1996) Adv. Drug Deliv. Rev. 18, 311-324.
[99] Meidan, V.M.; Alhaique, F.; Touitou, E. (1998) Acta Technologiae et Legis Medicament. 9(1), 1-6.
[100] Johnsen, S.G. Bennett, E.P.; Jensen, V.G. (1974) Lancet, 2, 1473Ð1475.
[101] Touitou, E. (2002) Expert Opin. Biol. Ther. 2, 723-33.
[102] Ainbinder, D. and Touitou E. (2005) Testosterone Ethosomes for enhanced transdermal delivery. Drug Delivery. 12, 297-303.
[103] Bruke R.E. and Fahn, S. (1985) Neurology. 35, 1066-1069.
[104] Touitou, E. Composition and methods for intracellular delivery. PCT/IL02/00516, 2002.
[105] Godin, B. and Touitou, E. (2005) Current Drug Delivery 2(3), 265-275
[106] Jain, S. Vesicular approaches for transdermal delivery of bioactive agent. Ph.D thesis, Dr. H.S. Gour University, Sagar, India, 2005.
[107] Tuting T.H.; Storkus, W.J.; Falo, J. (1998) J. Invest. Dermatol. 111, 183-188.
[108] Preat, V. and Dujardin, N. (2001) STP PharmaSciences 11, 57-68.
[109] Touitou, E.; Godin, B.; Dayan, N.; Vaisman, B. Intracellular delivery by cationic ethosomes not containing positively charged phospholipids: Characterization and intracellular delivery properties. Proceedings of the 6th PAT & 4th ICRS International Symposium, Eilat, Israel. 2001.
[110] Malfait, A.M.; Gallily, R.; Sumariwalla, P.F.; Anderkos, E.; Mechoulam, R.; Fieldman, M. (2000) Proc. Natl. Acad. Sci. 97, 9561-9566.
[111] Fang, J.; Hong, C.; Chiu, W.; Wang, Y. (2001) Int. J. Pharm. 219, 61-72.
[112] Kim, S. and Chien, Y.W. (1996) J. Control. Release. 40, 67-76.
[113] Jain, S. and Jain, N.K. (2004) In Progress in Controlled and Novel Drug Delivery Systems, Jain, N.K.; Ed., CBS publishers and Distributors, New Delhi, Vol. 1, pp 131-153,
[114] Corey, L. and Handsfield, H.H. (2000) J. Antimicrob. Chemother. 283(6), 791-794.
[115] Spruance, S.L. (1992) Semin Dermatol. 11, 200-206.
[116] Worral, G. (1991) Can. Fam. Physician. 37, 92-98.
[117] Huff, J.C.; Kreuger, G.C.; Overall, J.C.; Copeland, J.; Spruance, S.L. (1981) J. Am. Acad. Dermatol. 5, 550-557.
[118] Shaw, W.; King, M.; Best, J.M.; Banatrala, J.E.; Gibson, J.R.; Klaber, M.R. (1982) Br. Med. J. 291, 7-9.
[119] Fiddan, A.P.; Yeo, J.M.; Strubbings, R.; Dean, D. (1983) Br. Med. J. 286, 1699-701.
[120] Banga, A.K. and Chein, Y.W. (1993) Pharm. Res. 10, 697-702.
[121] Chetty, D.J. and Chien, Y.W. (1998) Crit Rev Ther Drug Carrier Syst. 15, 629-670.
[122] Dkeidek, I. and Touitou, E. (1999) AAPS Pharm. Sci.1, S202.
[123] Paolino, D.; Lucania, G.; Mardente, D.; Alhaique, F.; Fresta, M. (2005) J. Control. Release. 106, 99-110.
[124] Fu, Y.; Hsieh, J.; Guo, J.; Kunicki, J.; Lee, M.Y.; Darzynkiewicz, Z.; Wu, J.M.; Licochalcone, A. (2004) Biochem. Biophys. Res. Commun. 322, 263-270.
[125] Jain, S.; Umamaheswari, R.B.; Tripathi, P.; Jain N.K. (2003) Ind. J. Pharm. Sci. 65(3), 223-231.
[126] Touitou, E. and Godin, B.; Weiss, C. (2000) Drug Develop Research. 50, 406-415.
[127] Fry, D.W.; White, J.C.; Goldman, I.D. (1978) J. Anal. Biochem. 90, 809.
[128] New, R.R.C., (1990) In Liposomes: A practical approach, Oxford University Press, Oxford.
[129] Simonetti, O., Hoogstraate, J.; Bodde H.E. (1995) Arch. Dermatol. Res. 287, 465.
[130] Honeywell-Nguyen, P.L.; Graaff, D.; Anko, M.; Groenink, H.W.; Bouwstra, J.A. (2002) Biochim. Biophys. Acta. 1573, 130-138.
Authors
1Department of Pharmaceutical Sciences and Drug Research, Punjabi University, Patiala, Punjab, INDIA, 147 002.
2Department of Pharmaceutical Sciences, Dr. H.S. Gour University, Sagar (M.P.), 470003
*For correspondence
Dr. Subheet Jain
Department of Pharmaceutical Sciences and Drug Research
Punjabi University
Patiala (Punjab), India 147 002
Ph: +91-98158-99705
| Click on these buttons to visit our journals | ||||||||
| Psychiatry On-Line | Dentistry On-Line | Vet On-Line | Chest
Medicine On-Line | GP On-Line | Pharmacy On-Line | Anaesthesia On-Line | Medicine On-Line | Family
Medical Practice On-Line |
All pages copyright ©Priory Lodge Education Ltd 1994-2006.